Мышечная дистрофия Дюшенна
Невролог, нейрофизиолог, стаж - 38 лет;
Профессор неврологии, доктор медицинских наук;
Клиника восстановительной неврологии.Об авторе

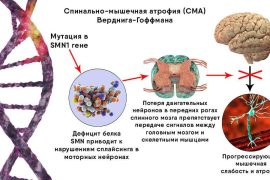
Существование генетических патологий, возникающих у людей только одного пола, — это результат мутаций в непарных хромосомах, возникающий, как правило, из-за матери, являющейся носительницей. Ярким примером такого заболевания является прогрессирующая миопатия Дюшенна. [1] Эта болезнь связана с нарушением в материнской X-хромосоме и проявляется только у мальчиков за редчайшим исключением.
ПМД Дюшенна проявляется в возрасте 3-5 лет поражением мышц таза и бёдер, постепенно распространяется по спине, по конечностям. К 15 годам больной уже будет окончательно обездвижен и не сможет жить без аппарата ИВЛ.
Это тяжёлая форма миодистрофии, которая, постепенно распространяясь по всем мышцам организма, приводит к выраженной деформации скелета. На последних стадиях происходит поражение сердечной мышцы и полное прекращение дыхания. Люди, страдающие этим заболеванием, не доживают и до 25 лет даже с постоянной медицинской помощью. При этом частотность болезни 1 человек на 4-5 тысяч мальчиков и постепенно возрастает.

Заболевание было описано впервые французским неврологом Дюшенном ещё в 1853 году. Уже тогда врач заметил, что миопатия Дюшенна у девочек практически не встречается, но определить, что её вызывает, не мог, так как наука ещё не знала о генетическом факторе.
В большинстве случаев развитие болезни провоцирует мутация в гене дистрофина, расположенном в X-хромосоме, передающейся от матери. Только одна X-хромосома и стала причиной того, что болезнь развивается у мальчиков. У девочек крайне редко его тоже выявляют, но в значительно более лёгкой форме. Для них оно не смертельно и проявляется небольшой слабостью мышц и заболеваниями миокарда.
Однако далеко не всегда в развитии мышечной дистрофии виноват генный материал женщины:
- Миопатия Дюшена может быть вызвана и другими видами мутаций, передающихся плоду.
- В 30% случаев заболевание развивается не из-за переданного от матери «сломанного» генного материала, а из-за специфической мутации, появившейся при развитии плода.
Симптомы

Болезнь проявляется постепенно, но уже в первые годы жизни даёт о себе знать. Внимательный родитель или врач увидит отклонения в развитии с самых первых месяцев. Официально первые симптомы прогрессирующей дистрофии мышц фиксируются с 1 до 5 лет, но на самом деле первые предвестники видны даже раньше. [2]
Мышечная слабость начинает активно становиться заметной уже в 3-4 года. Сначала появляется быстрая утомляемость, ребёнок устаёт намного быстрее сверстников при подъёме по лестнице или при прогулках на длинные расстояния. Его походка постепенно меняется, становясь менее устойчивой, чуть раскачивающейся, утиной. Ещё одна отличительная черта – необычный способ подниматься из положения сидя на корточках. Малыш помогает себе руками, упирающимися в тело, словно взбирается по лесенке.
Со временем проблема будет усугубляться и проявятся и другие симптомы:
- Псевдогипертрофия икроножных мышц.
- Формирование «осиной» талии.
- Лопатки становятся выступающими, сильно отстающими от спины.
- Выпадают сухожильные рефлексы начиная с коленных. При этом Ахилловы и карпорадиальные рефлексы сохраняются достаточно долго.
- Ретракции сухожилий.
- Мышечные контрактуры.
- Значительное искривление позвоночника.
- Деформация костей стопы.
- Изменённая форма грудной клетки.
- Сердечно-сосудистые заболевания.
Осложнения
Впервые прогрессивная мышечная дистрофия манифестирует в возрасте 3-5 лет. Уже через несколько лет, к 7 или 10 годам симптомы проявляются достаточно сильно, чтобы мешать полноценному передвижению. К 12 годам в большинстве случаев пациенты утрачивают способность самостоятельно передвигаться. К 15 годам мальчика ждёт полный паралич без возможности самообслуживания даже на минимальном уровне.
Диагностика
Заболевание относится к действительно сложным и достаточно редко встречающимся, имеет симптоматику, схожую с множеством других болезней, поэтому диагностику необходимо проводить особенно тщательно и всеобъемлюще. Только проанализировав результаты исследований разного типа, врач сможет с полной уверенностью поставить диагноз и назначить поддерживающее лечение. [3]

При диагностике в обязательном порядке проводятся:
- Генетическое обследование. Сначала врач-генетик проводит обследование, помогающее выявить наличие синдрома у других родственников по материнской линии, чтобы понять, могла ли мать передать заболевание сыну. Проводится тест ДНК, позволяющий выявить наличие мутации гена, которая и приводит к развитию мышечной дистрофии. Однако стоит понимать, что его результат не всегда 100%, так как выявление точечных мутаций, тоже способных спровоцировать развитие болезни, в обычных лабораториях практически не делают.
- Биопсия мышечной ткани приходит на помощь, если ДНК анализ не выявил мутации. Материал, забранный из мышцы, исследуется на предмет разнокалиберности и некроза миоцитов. Также может быть выявлено отсутствие в тканях дистрофина.
- Электронейро- и электромиография помогает определить, сохранилась возможность проведения сигнала по нервным волокнам. Это показывает, что поражены именно мышцы, а не нервные ткани.
- Дополнительные исследования помогают выявить, какие ещё изменения в организме происходят под воздействием развивающегося заболевания. В обследование входит обязательный осмотр кардиологом и проведение ЭКГ, рентгена позвоночника и эхокардиография. Дополнительно назначаются обследования специалистов узкой направленности в зависимости от состояния пациента.
Лечение
Стандартная терапия

Подбирая методы лечения для больных со столь сложным диагнозом, врач разрабатывает индивидуальную терапию, сочетая методы, которые позволят ослабить симптомы и замедлить развитие патологии с учётом индивидуальных особенностей каждого пациента. Для этого учитывается результат диагностики и анамнез не только самого ребёнка, но и его родителей.
Обычно в терапию входят:
- Приём лекарственных препаратов. На данный момент основу лечения мышечной дистрофии Дюшенна составляет регулярный приём кортикостероидов. Они замедляют процесс ослабления мышц, поэтому их прописывают как двигающимся, так и утратившим такую возможность пациентам. Препараты считаются опасными из-за немалого количества побочных эффектов, поэтому ребёнку требуется постоянный надзор врача, регулярные осмотры и корректировки дозы и схемы приёма препаратов.
- Метаболическая терапия. Является поддерживающей и снижающей побочные эффекты от приёма глюкокортикостероидов. Её главная задача – улучшить обменные процессы, затрагивающие скелетную мускулатуру, печень, сердце, кости. В неё входит приём группы витамином, обеспечивающих исправную работу обменных процессов.
- Физическое лечение является одним из важнейших элементов, позволяющих максимально продлить период двигательной активности пациента. В ходе неё используется специальный комплекс упражнений ЛФК, лечебное плавание, растяжки, массажи. Физиотерапия в комплексе процедур ТМС, БОС, КВЧ и т.д. Дополнительно используются вспомогательные элементы: специальные шины и вертикализаторы.
- Респираторная поддержка. Дыхательная функция особенно страдает в последний период развития болезни. Сначала ЖЕЛ падает до 40%, и пациенту будет требоваться подача кислорода в период сна для поддержания дыхания. Затем человек переходит полностью на ИВЛ без возможности самостоятельного дыхания. Современные аппараты работают от батареек, что позволяет сделать их портативными и прикреплять к инвалидному креслу, не лишая пациента способности к передвижению. [4]
Экспериментальные методики
До сих пор врачи и учёные всего мира пытаются найти действенный вариант лечения прогрессирующей мышечной дистрофии. Самым перспективным вариантом на сегодня является разработка лекарств, которые должны будут либо заместить дистрофин, либо восстановить его синтез в организме до нормального уровня. На данный момент в мире известно несколько препаратов, которые показывают хорошие результаты при условии пожизненного приёма.

В России в 2020 году прошёл сертификацию первый таргетный препарат, созданный для полноценного лечения синдрома Дюшенна – аталурен. Однако он подходит всего 10% пациентов, у которых заболевание было спровоцировано нонсенс-мутацией. Приём лекарственного средства помогает возобновить естественный синтез белка и остановить дальнейшую деградацию мышечных тканей. Ещё несколько препаратов, обещающих аналогичный результат, в России пока не зарегистрированы.
Профилактика
Защитить уже рождённого ребёнка от наследуемого синдрома мышечной дистрофии невозможно. Девочка станет просто носительницей с возможностью передачи болезни своим детям и последующим её развитием у мальчиков.
При подготовке к беременности пара должна пройти обследование у генетика и собрать необходимый тщательный анамнез о своих семьях и случаях развития миодистрофии. После зачатия в период вынашивания плода также проводится консультация с генетиком и пренатальная ДНК-диагностика, помогающая определить возможность развития патологии ещё на раннем сроке.

Список использованной литературы:
- ^ Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. “Duchenne muscular dystrophy.” Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3. PMID: 33602943; PMCID: PMC10557455.
- ^ Chelly J, Desguerre I. “Progressive muscular dystrophies.” Handb Clin Neurol. 2013;113:1343-66. doi: 10.1016/B978-0-444-59565-2.00006-X. PMID: 23622359.
- ^ Falzarano MS, Scotton C, Passarelli C, Ferlini A. “Duchenne Muscular Dystrophy: From Diagnosis to Therapy.” Molecules. 2015 Oct 7;20(10):18168-84. doi: 10.3390/molecules201018168. PMID: 26457695; PMCID: PMC6332113.
- ^ Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM; DMD Care Considerations Working Group. “Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management.” Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5. Epub 2018 Feb 3. PMID: 29395990; PMCID: PMC5889091.